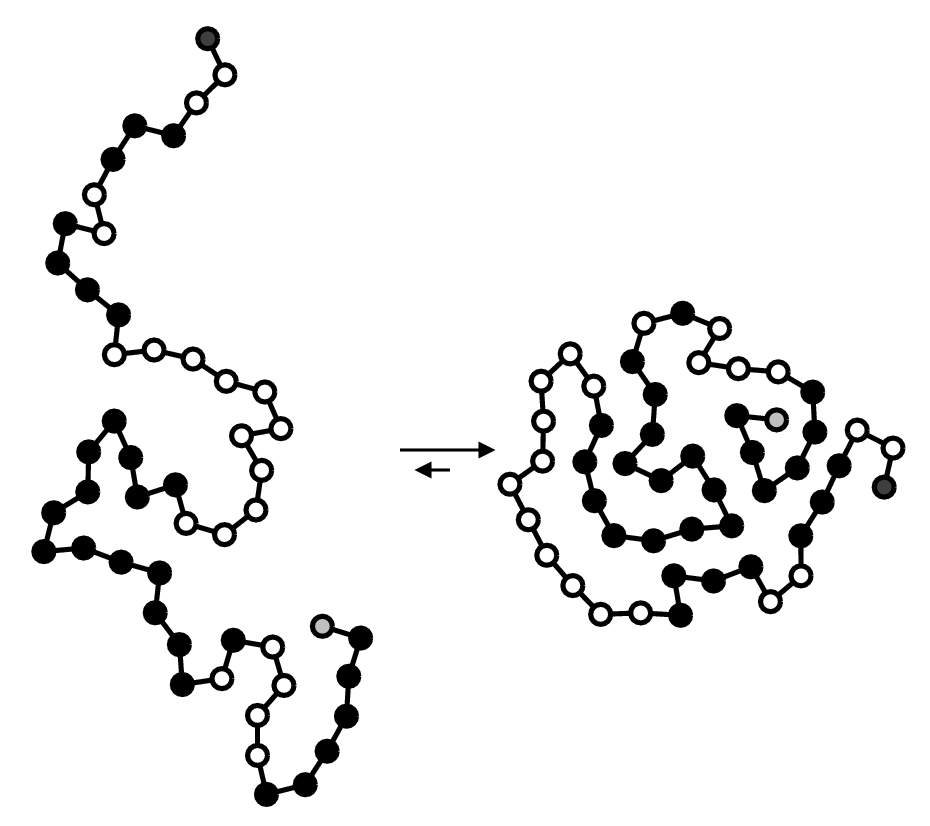
Epistemic status: I have no expertise, but below is my best guess from what I have read so far, mostly relying on AlQuraishi’s excellent essay. The first half of this post will be kind of a literature review on the protein structure prediction, and the second half is a bit of a rehash of AlQuraishi’s essay, so post will serve as a companion piece of sorts to help you understand the background of the problem better if you’re a layman like me.

DeepMind, Google’s premier AI offshoot, predicted protein structures so impressively with their AlphaFold 2 (AF2) in the Critical Assessment of Techniques for Protein Structure Prediction (CASP), the biennial international competition of at predicting protein structure, that the organisers declare the protein structure prediction problem for single protein chains to be solved.
 They see me foldin’ they hatin’
They see me foldin’ they hatin’
Is the hype for real?1 Mohammed AlQuraishi, a computational biologist at Columbia University and CASP participant, said,
“In my read of most CASP14 attendees (virtual as it was), I sense that [the protein structure prediction problem for single protein chains was solved] was the conclusion of the majority. It certainly is my conclusion as well.”
As if it wasn’t hyperbolic to describe this moment to be protein structure prediction’s ImageNet moment like some have, AlQuraishi said,
“It is more akin to having the ImageNet accuracies of 2020 in 2012! A seismic and unprecedented shift so profound it literally turns a field upside down over night.”
- But Why?
- A Very Hard Problem
- So You Want To Predict Protein Structures
- CASP
- Top Dogs
- AlphaFold 2: Electric Boogaloo
- How Did DeepMind Do It?
- Why DeepMind?
- Wait, So Is The Protein Folding Problem Solved?
- What Are The Implications?
- Will It Revolutionise Drug Discovery?
- Endnotes
But Why?
The central dogma of molecular biology states that the flow of genetic information within a biological system cannot be transferred back from protein to either protein or nucleic acid per Francis Crick’s original statement in 1957 . The more popular version as stated by James Watson in 1965 is often known as “DNA makes RNA, and RNA makes protein (amino acid sequence)”
However, we still can’t tell what the function of the protein is until we learn about its structure because structure is function: how a protein works and what it does is determined by its native (properly-folded) conformation (3D shape).
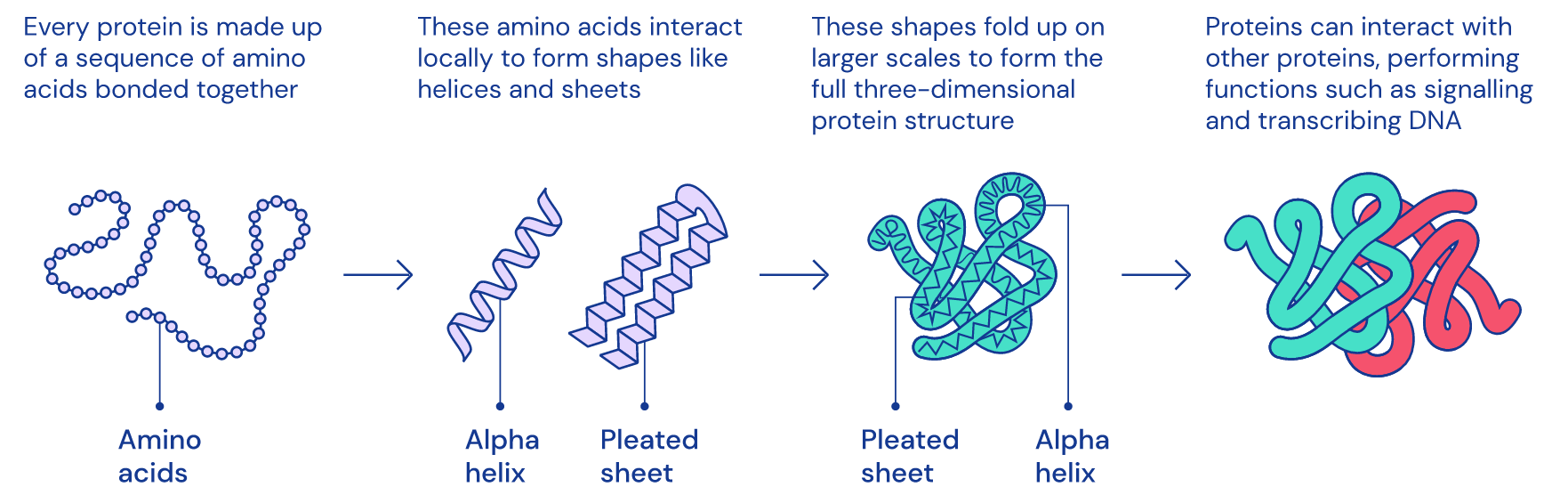 1D amino acid sequence fold into 3D blobs
1D amino acid sequence fold into 3D blobs
To determine the structure of a protein, the predominant approach is to look at its conformational changes during gradual unfolding or folding with experimental techniques (e.g. X-ray crystallography, nuclear magnetic resonance spectroscopy, and more recently cryogenic electron microscopy a.k.a. cryo-EM).
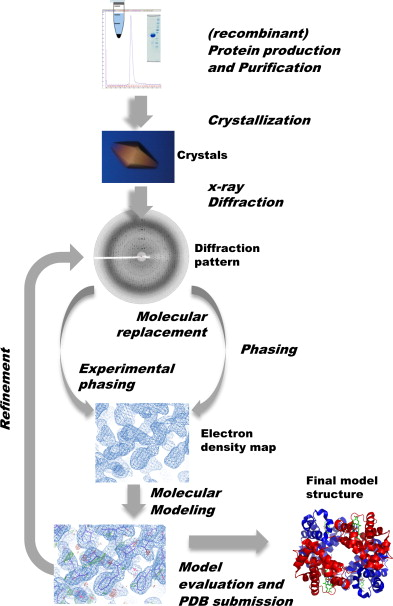
Steps of X-ray crystallography
As these methods depend on extensive trial and error, it can take years of painstaking and laborious work per structure, and require the use of multi-million dollar specialised equipment. For example, it takes about a year and $120000 to obtain the structure of a single protein with X-ray crystallography. Worse, they don’t work for all proteins. Notably, membrane proteins (e.g. the ACE2 receptor that SARS-CoV-2 binds to) are difficult to crystallise, yet they represent the majority of drug targets.
This means that we have only determined the structure of a tiny percentage of proteins we’ve sequenced: there are more than 200M protein sequences in the Universal Protein Reference Clusters (UniRef), but only 170k structures in the Protein Data Bank (PDB), and the gap is only widening as genome sequencing has been improving at a breathtaking rate.
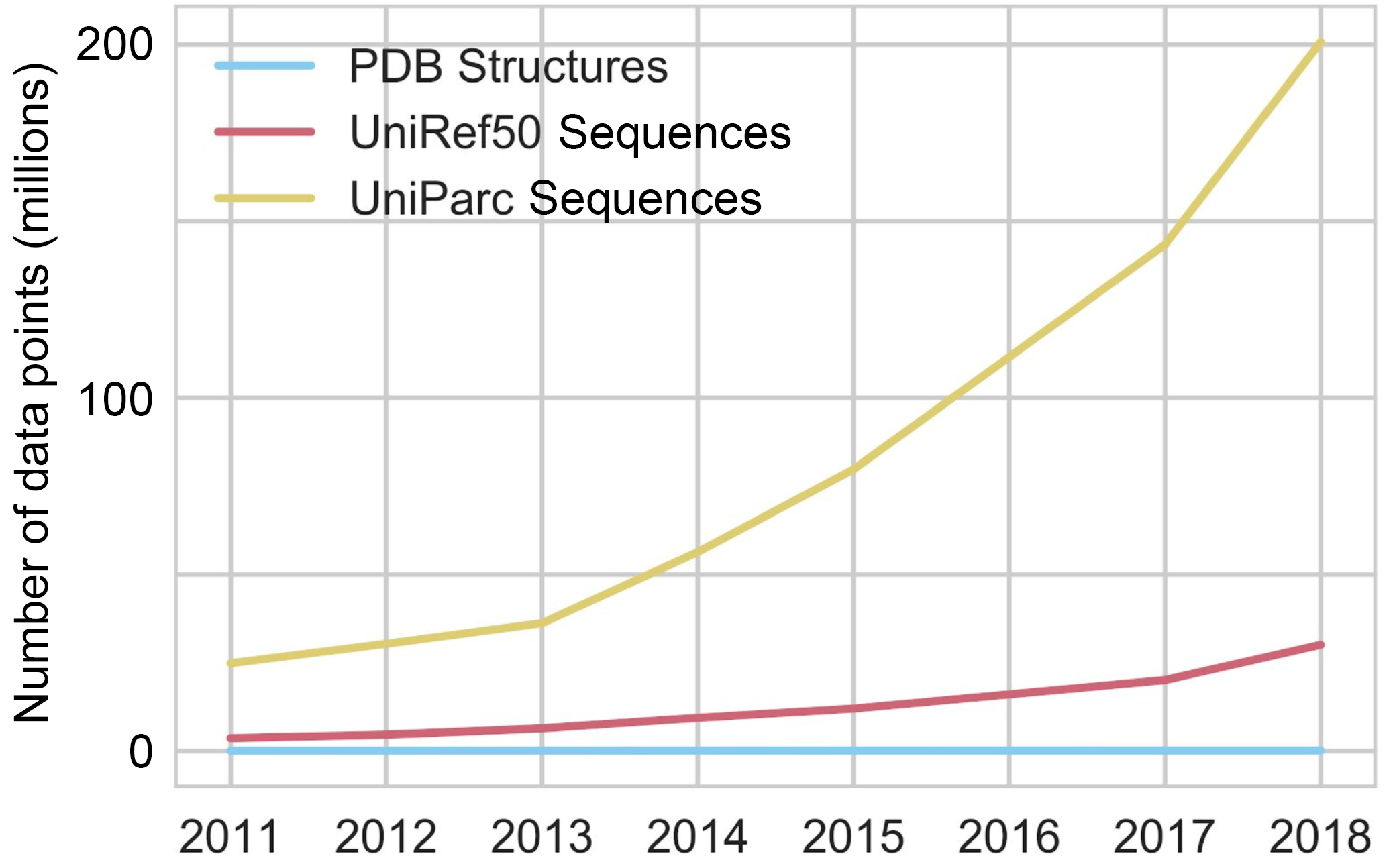 Sequencing will continue until morale improves.
Sequencing will continue until morale improves.
When Christian Anfinsen won the 1972 Nobel Prize in Chemistry for Anfinsen’s dogma (a.k.a. the thermodynamic hypothesis), he postulated that to determine a protein’s native structure, all the information you need is in its amino-acid sequence. So what if we could determine the 3D structures of proteins much faster and much faster by folding amino acid sequences in silico?
In the 6 decades since the 1962 Nobel Prize in Chemistry was awarded to Max Perutz and John Kendrew for founding the field of structural biology, the protein-folding problem actually came to be three main questions:
-
The physical folding code: How is the 3D native (properly-folded/functional) structure of a protein determined by the physicochemical properties that are encoded in its 1D amino-acid sequence?2
-
The folding mechanism: A polypeptide chain has an almost unfathomable number of possible conformations. How can proteins fold so fast?3
-
Predicting protein structures using computers: Can we devise a computer algorithm to predict a protein’s native structure from its amino acid sequence?
So whenever you hear people shout that DeepMind has solved the grand challenge of the protein folding problem, they’re really only referring to the protein structure prediction problem.
A Very Hard Problem
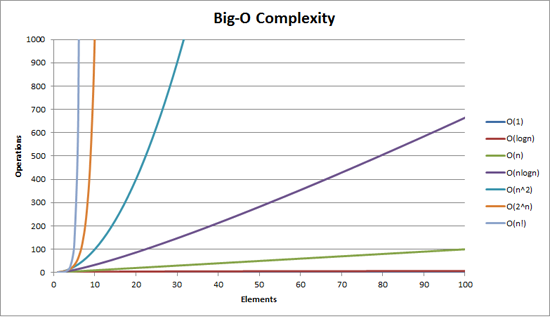
So you want to predict protein structure in silico, the obvious way is to simulate physics. With molecular dynamics (MD), you can try to model the forces on each atom, given its location, charge, and chemical bonds, then calculate accelerations and velocities and evolve the system step by step. However, a typical protein has hundreds of residues (amino acids), which means thousands of atoms. Then there are the like 30k more atoms to simulate because of surrounding water molecules. And there are electrostatic interactions between every pair of atoms. So naively that’s ~450M pairs.
Simulating physics has the Big-O complexity of O(n2), shown as the turquoise line in the graph below. (There are smart algorithms to make this O(N log N).) More like Big Oof complexity because you end up needing to run for something like 109 to 1012 time-steps4.
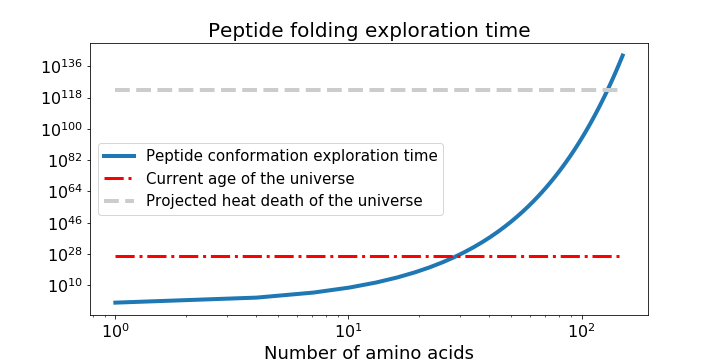
Just when you’re questioning your hubris, you remember that Anfinsen’s dogma5 also postulates that a protein’s native structure has the minimal global free energy, so maybe you can use the same MD model to calculate what structure minimises free energy the most. You do this by sampling the configuration space (set of all possible conformations), guided by scoring functions (potential-energy minimising) to produce a large set of candidates, then selecting the better ones with more scoring functions, before high-resolution refining. But as Cyrus Levinthal noted in 1969, the number of possible conformations is astronomically large, like 10300 (!) large. It will take you more time than the age of the universe to solve the 2 main problems of calculating the protein free energy and finding the global minimum of that energy. 2D and 3D mathematical modelling of protein folding as a free energy minimisation problem is NP-hard!
So You Want To Predict Protein Structures
3.1. Conformation initialisation
Protein structure prediction methods are categorised into template-based modelling (TBM) and free modelling (FM) with the key difference in how they arrive at the initial conformation. In fact, both categories of prediction methods make use of the structural information from previously solved structures in the PDB to after conformation initialisation, as the basic observation is that similar sequences from the same evolutionary family often adopt similar protein structures.
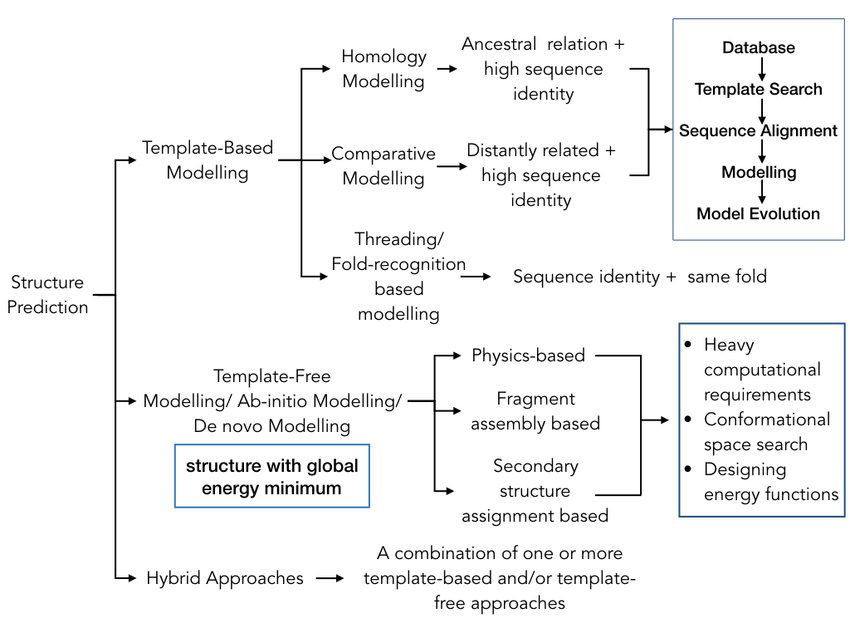
The computational-intensive problems of calculating the protein free energy and finding the global minimum of that energy can be partially bypassed in TBM, in which the search space is pruned by the assumption that the protein in question adopts a structure that is close to the experimentally determined structure of another homologous protein. The steps in the canonical procedure of the TBM are:
-
finding known structures (templates) in the PDB related to the sequence to be modeled (target);
-
aligning the target sequence to the template structure;
-
building structural frameworks by copying the aligned regions or by satisfying the spatial restraints from templates;
-
constructing the unaligned loop regions and add side-chain atoms;
-
evaluating the model using a variety of internal and external criteria.
On the basis of shared sequence identity, TBM can be classified into:
-
Homology Modelling (HM): for target proteins that have homologous proteins with known structure (usually from the same family/share ancestry), compares 1D target sequence to 1D template sequence;
-
Comparative Modelling (CM): for target proteins that have no identified evolutionary relationship with the template but only shared sequence similarity, compares 1D target sequence to 1D template sequence
-
and Protein Threading (a.k.a. fold-recognition): for target proteins that do not have homologous proteins with known structure, but have the same fold as known structures, compares 1D target sequence and 3D template structure by “threading” (i.e. placing, aligning) each amino acid in the target sequence to a position in the template structure and evaluating how well the target fits the template
HM and CM are effective as long as the target sequence shares at least 25-30% identity with the template. For any target lower than that, threading is used.
If you think TBM is too easy, you can try FM (a.k.a. template-free or ab initio or de novo simulation), where no solved homologue of the protein target is used, so you have to explicitly resolve the computationally-intensive problems. Sometimes you don’t really have a choice because a significant amount of target sequences aren’t homologous with well-studied protein families. There are:
-
Physics-based approaches: good luck
-
Fragment-based approaches (FBA): by far the most successful, first predict the backbone dihedral angle, solvent accessibility, contact map or secondary structure segments of the target sequence, then replace the sequence with fragments cut from known structures in the PDB based on those predictions
-
Secondary-structure-elements- (SSE) based approaches: assembling the core backbone of the protein with an exception of loop regions leading to model refinement protocols
Whether you’re doing TBM or FM or a hybrid of both, you will be mixing and matching residues, which makes the side chain conformations from template unusable, so the initial reduced conformation you get only has the backbone carbon atoms (Cα). For example, each residue can be represented just by its Cα-atom and the virtual center of its side chain, or the entire backbone conformation can be represented by a series of dihedral angles, which allows you to cut down a lot of time.
3.2: Conformational search and structure selection
Searching through the conformation space requires a “force field)” depicting the protein conformational energy landscape, where all conformations correspond to a point on the energy landscape (the native conformation should be the lowest point).
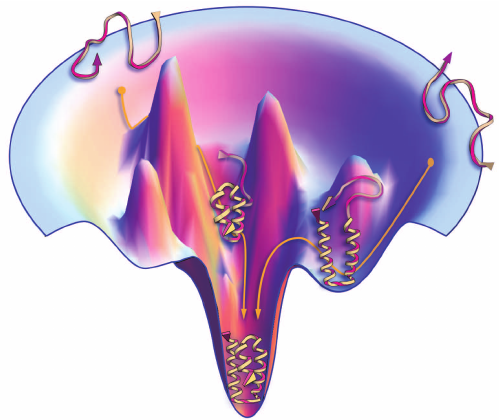
Protein folding guided by funnel-shaped energy landscape
Proteins fold due to atomic interactions, so ideally force fields should be constructed based on quantum mechanics. Alas, until quantum computers materialise, you can only construct force fields based on classical physics that consist of the bond-stretching energy, the angle bending energy, the angle torsional energy and other energies for nonbonded interactions. Due to computational limits, these physics-based energy functions are often inaccurate for describing the complicated atomic interactions and have poor performances in protein structure prediction.
On the other hand, you can use force fields constructed based on information such as distance, dihedral angles, solvent accessibility, and side chain orientation of the many experimentally-determined structures in the PDB. This is where machine learning methods like hidden Markov models, artificial neural networks, and support vector machines come to play. This unfortunately means that such knowledge-based energy functions are constructed with “black boxes” which cannot help you understand the nature of the protein folding dynamics.
 Overview of Alpha Fold the first
Overview of Alpha Fold the first
In practice, most structure prediction methods employ both physics-based and knowledge-based energy functions, with which you have to find a proper way to search for the minimal global free energy conformation of the target protein. One way is MD, but if you recall, that’ll take more time than the age of the universe. Worse, to make statistically valid conclusions from the simulations, the time span simulated should match the kinetics of the natural process, otherwise it’s like making conclusions about how a human walks by only looking at less than one footstep. The time step of MD is generally in the order of femtosecond (10−15 s), while the time required for protein folding is usually in milliseconds (10−3 s). Yeah that’s right, as if MD doesn’t already take long enough, each time step should be a trillion times slower.
A much faster method for conformation search is based on Monte Carlo simulation, which randomly changes the conformation based on various movements designed beforehand that includes the shifts and rotations of structural segments as well as the position changes of a single atom. These movements can be spatially continuous, or confined to a cubic lattice system, which can dramatically reduce the conformation space.
Whether you’re using MD or Monte Carlo simulations, your force field will not be good enough so the conformations are often trapped at the local minimal state, even when the global minimal state is identified. Hence, the common procedure during simulation is to regularly output lower energy intermediate structures. In fact, there is a specific category in CASP for assessing the methods of structural quality assessment to check which of the many conformations you end up with is actually the native one. You can perform structure selection with energy functions much more complicated than the ones you use for conformation search because you have a lot fewer conformations to choose from.
3.3: All-atom structure reconstruction and structure refinement
Now you end up with just one or several reduced structural models with only the Cα-atoms, so you have to rebuild the backbone atoms (carbon, nitrogen, oxygen) and the side chain for every residue using fragments cut from experimental structures.
Unfortunately the all-atom model isn’t very good due to defects of the force field, conformational search or all-atom reconstruction, so you need to slightly tweak the local details of the backbone conformation with more MD or Monte Carlo simulations. In fact, there is also a specific category for model refinement since CASP7.
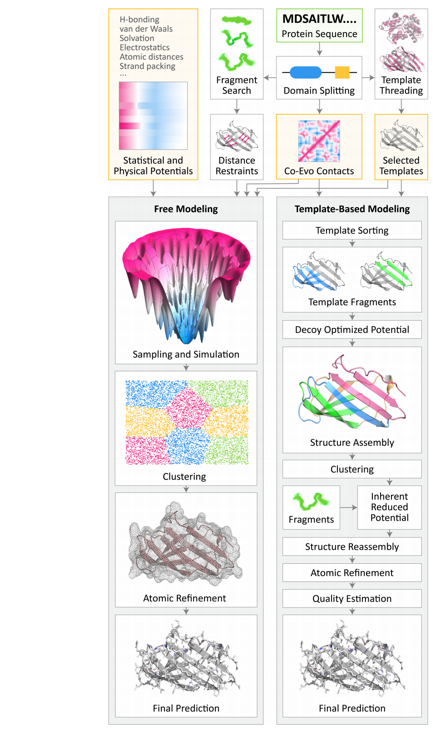
Hopefully this flowchart of TBM and FM now makes sense.
CASP
To compare the different protein structure prediction approaches, we can look at their performances in the Critical Assessment of Techniques for Protein Structure Prediction (CASP), a double-blinded “world championship” held every 2 years since 1994, where ~100 teams try to predict ~100 domains of protein native structures that have been recently solved (overwhelmingly by the US Protein Structure Initiative) and temporarily withheld from PDB.
There are many categories such as multimeric targets (for protein complexes), and refinement targets, but the two categories relevant to AlphaFold are tertiary structure prediction and contact (or distance) prediction.
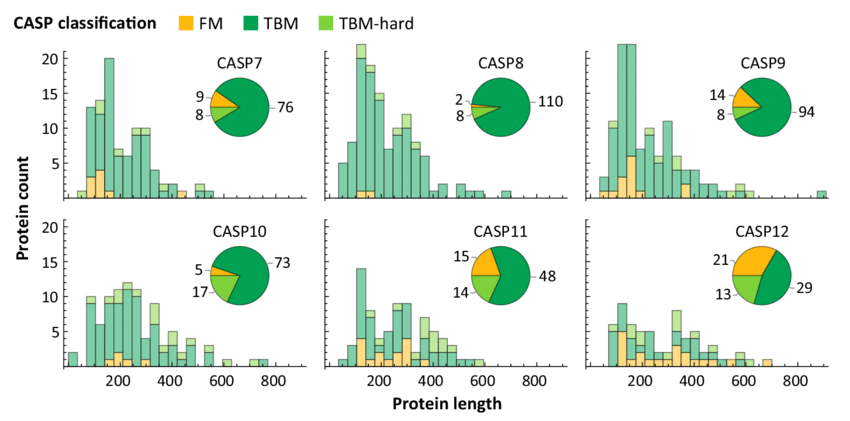
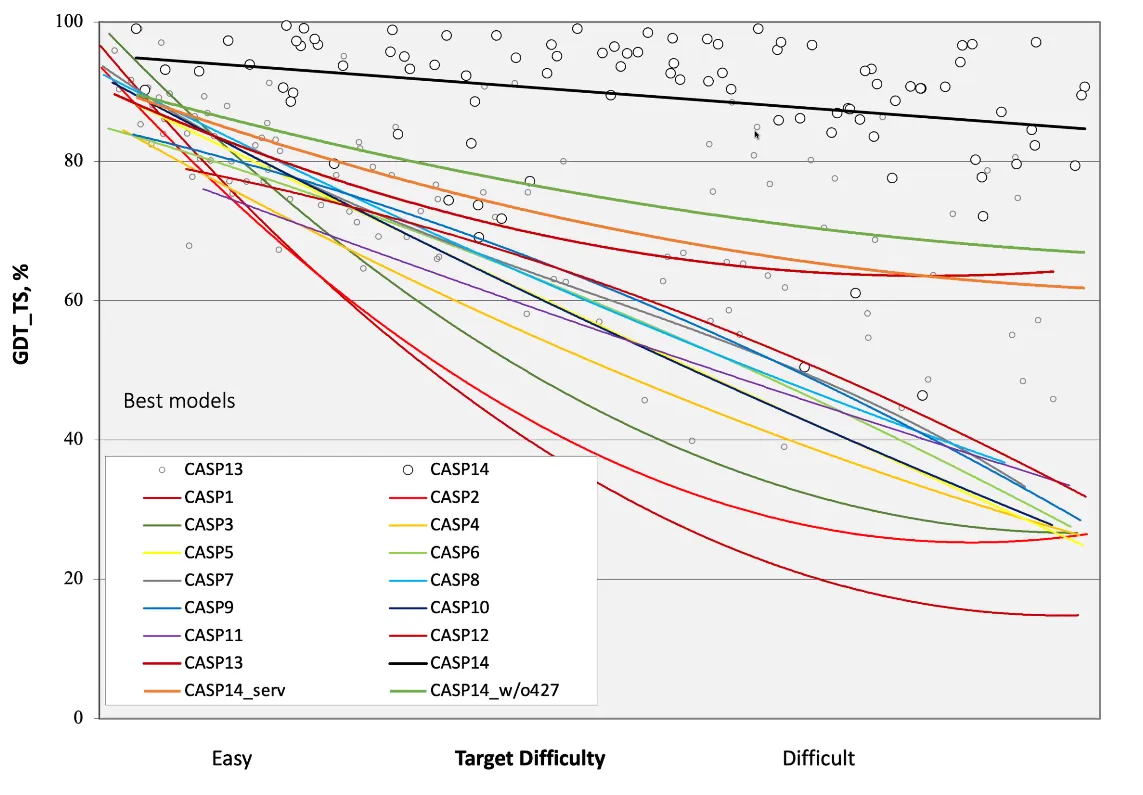
CASP structures have on average grown in length, although the vast majority of structures remain shorter than 1,000 residues:
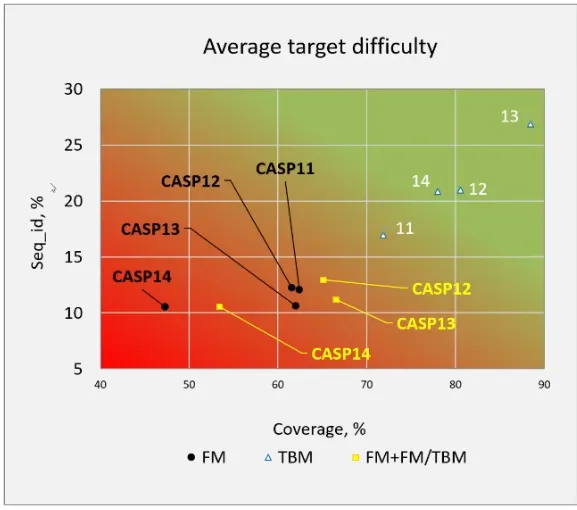
The average difficulty of the targets in CASP has also increased. In fact, the targets for CASP14 were the most difficult to date:
There are 2 types of predictors: the human, and the server. For each protein target, the former has 3 weeks and can make use of all the server results and any available resources, while the latter only has 3 days and cannot see the result from other servers. As it is “openbook” for humans, they can easily beat most of the servers by simply doing a consensus analysis (majority voting) on all server predictions, or straight up just copying the results from the best servers.
One metric used by CASP is the global distance test “total score” (GDT_TS) from 0-100 (the higher the better). It is calculated by superimposing the predicted tertiary structure onto the experimentally determined (via X-ray crystallography or protein NMR) tertiary structure, and adding the percentage of alpha carbons (first carbon atom that attaches to a functional group) that fall within cutoffs of 1, 2, 4, and 8 Å (10−10 m), then divide the sum by 4.
What’s a good score? Random predictions give around 20 GDT; getting the gross topology right gets one to about 50-60 GDT; accurate topology is usually around 70-80 GDT; and when even the side-chain conformations are correct, GDT_TS begins to climb above 90 GDT; and anything above 95 GDT is within experimental accuracy. According to John Moult, one of the founders of CASP who declared AF2 has solved protein structure prediction for single protein chains, around 90 GDT is informally considered to be competitive with results obtained from experimental methods.
Another metric that is more commonly used by the broader biology community is the root-mean-square deviation (RMSD). It is calculated by taking the root mean square of the distance between alpha carbons of the superimposed structures. RMSD is sometimes expressed as RMS_CA or Cα-RMSD, as it only looks at alpha carbons, while RMS_ALL is used when looking at all atom positions (the other 90% of the protein).
What is a good RMSD? There doesn’t seem to be a strict answer and it’s a bit like comparing apples (accuracy) to oranges (resolution), but in silico models aim for a Cα-RMSD smaller than 2.5 Å (for reference X-ray crystallography is high resolution at < 3 Å).
In the first ten years of CASP, the technique that made the most rapid progress was TBM. Progress has slowed in the following 8 years when many attempts to improve on the best template ironically turned it into a worse model. Fortunately, there have been substantial improvements recently with average precision increasing from 27% in CASP211 (2014) to 47% in CASP12 (2016), in part due to improved understanding of the energetics of protein folding but also largely by taking advantage of the growing databases.
As for FM, The first time anyone got it remotely right was around CASP3 (1998), and the subsequent progress has mostly just been on small, single-domain proteins, but hey at least there is progress.
One of the factors that lead to the recent breakthrough of FM is residue‐residue coevolutionary analysis as demonstrated in 2014 CASP11 and 2016 CASP12. You can narrow down the search space when you realise that when one amino acid mutates to be positively charged, another amino acid must mutate to be negatively charged for the two amino acids to keep in contact during evolution.
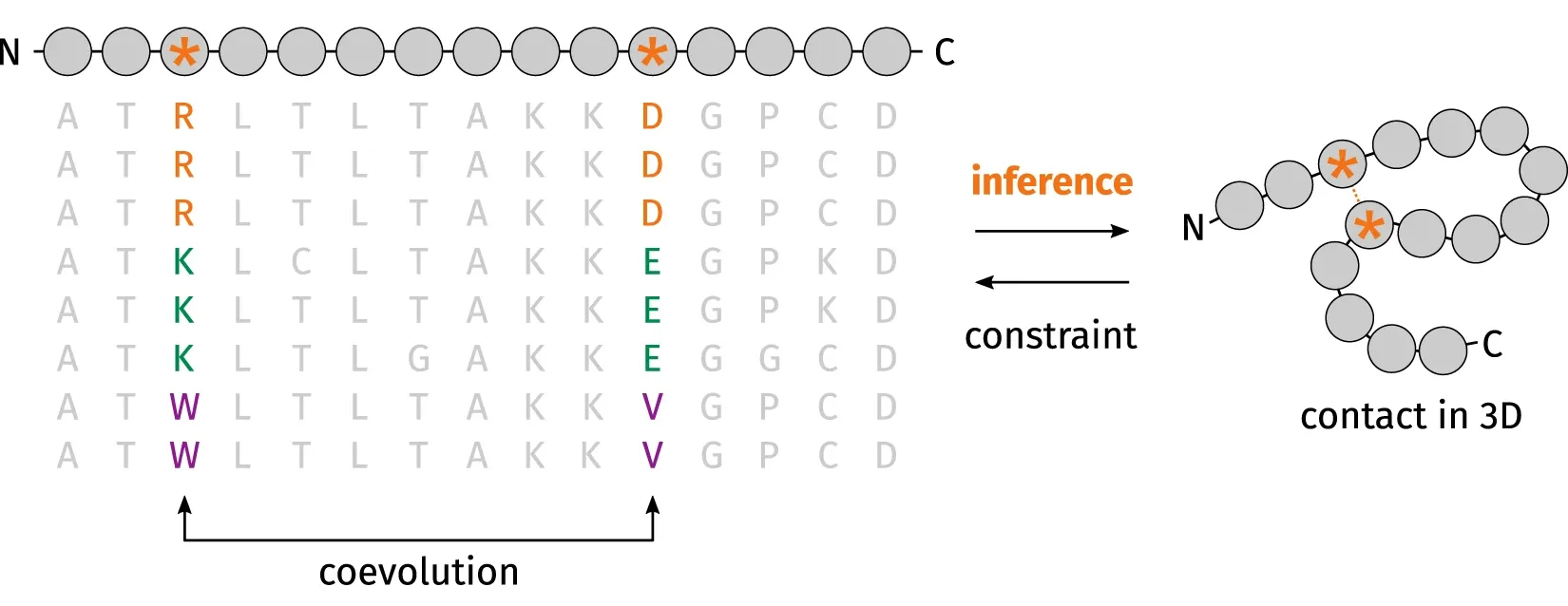
Another factor is deep learning which was introduced into the field in 2012 and demonstrated as the best method for protein contact prediction in 2012 CASP10. There was significant improvement in 2016 CASP 12 where convolutional neural networks, residual networks, coevolutionary analysis, fragment assembly, and distance geometry were combined to build protein structural models from scratch.
Top Dogs
One of the consistently top-ranking servers in CASP is I-TASSER (as Zhang-Server) developed by the Yang Zhang Lab at the University of Michigan. It uses threading and Monte Carlo simulations based on fragments.
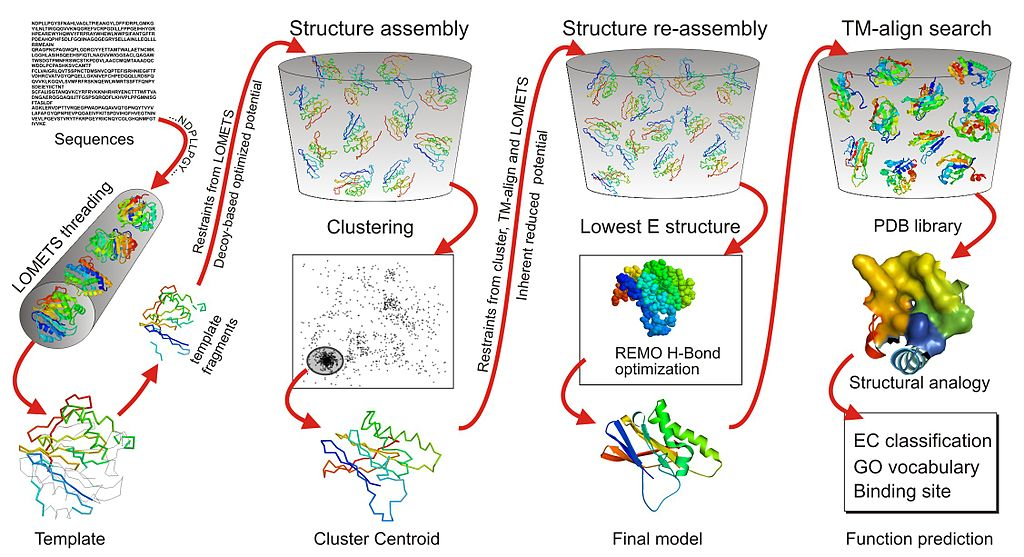
Another top-ranking server is Rosetta developed by the David Baker Lab at the University of Washington. It is optimised for predicting the tertiary structure of single protein domains, with the first step usually being domain parsing (or domain boundary prediction) to first split a protein into potential structural domain using Monte Carlo simulation. After the structures of individual domains are predicted, they are docked together in domain assembly to form the final tertiary structure. Rosetta was released as Rosetta@home to use idle computer processing resources from volunteers’ computers to perform calculations. Baker lab also released Foldit, an online protein structure prediction game based on the Rosetta platform. As of March 28 2020, Rosetta@home had 54,800 computers providing an average performance of over 1.7 PetaFLOPS (1015 FLOPS). Despite huge computing power even back in 2007, it took almost 2 years and 70,000 home computers to predict the tertiary structure of the protein T0283 from its amino acid sequence. Rosetta@home’s highest achievement was correctly predicting the structure of a 256 amino acid long sequence in CASP11.
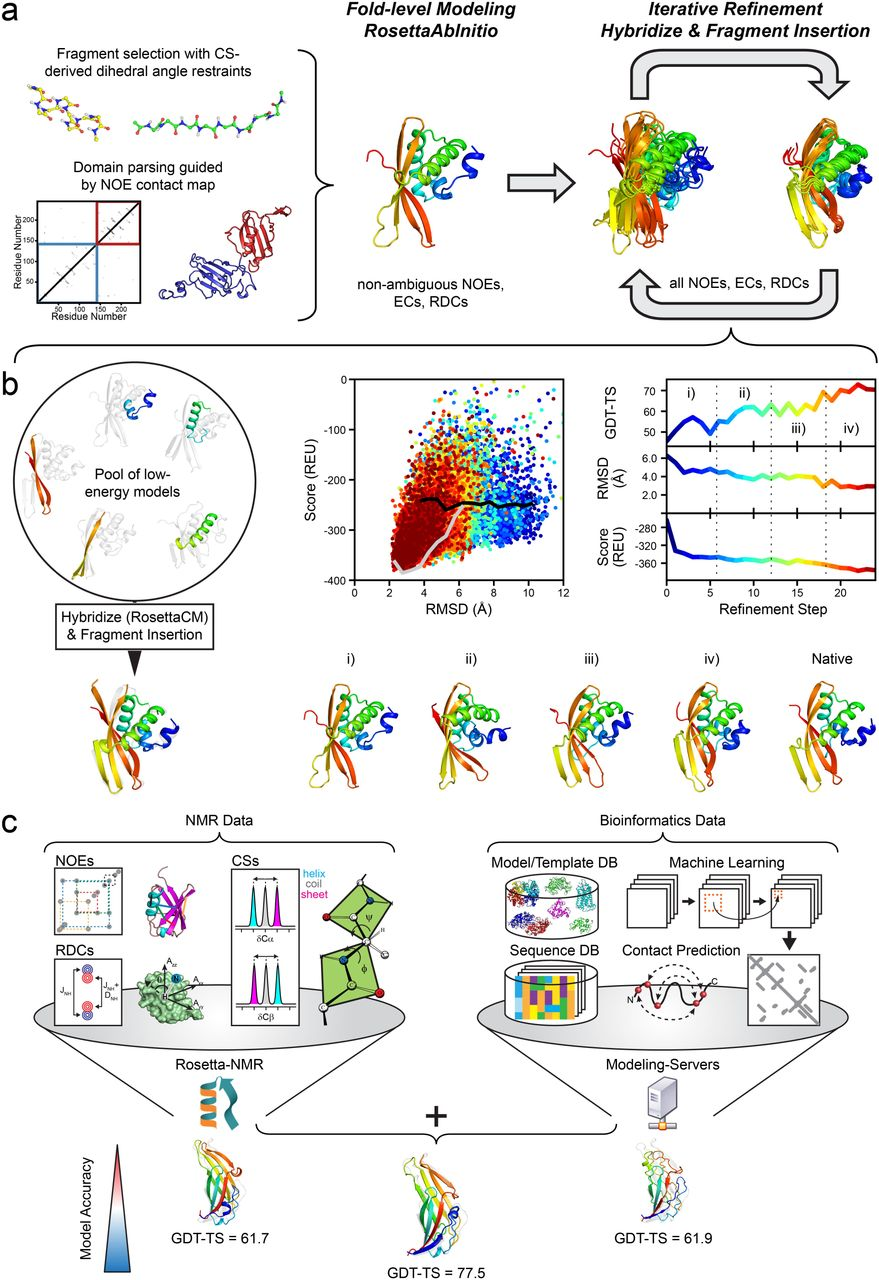 Overview of Rosetta’s protocol in the CASP13 NMR-assisted structure prediction category.
Overview of Rosetta’s protocol in the CASP13 NMR-assisted structure prediction category.
What about Stanford’s Folding@home (FAH)6? It’s probably the better known distributed computed project. The conformational states from Rosetta’s software can be used to initialize a Markov state model as starting points for FAH simulations. FAH almost exclusively uses all-atom molecular dynamics models to understand how and why proteins fold. As far as I know, it has never competed in CASP.
DeepMind debuted in CASP13 (2018) with Alpha Fold and came first in overall performance with a sum Z-score (the difference of a sample’s value with respect to the population mean, divided by the standard deviation) of 128.07. The top groups, especially the Baker and Zhang groups, are often running neck and neck, so DeepMind really proved themselves to be the α-ML by doing the best for 1/3 of targets. It was a bit overhyped though, as the result is ultimately within the bounds of the usual expectations of academic progress, albeit at an accelerated rate. The data shows that it often outperforms everyone else, but other times it just wanders off (which happens to every program).
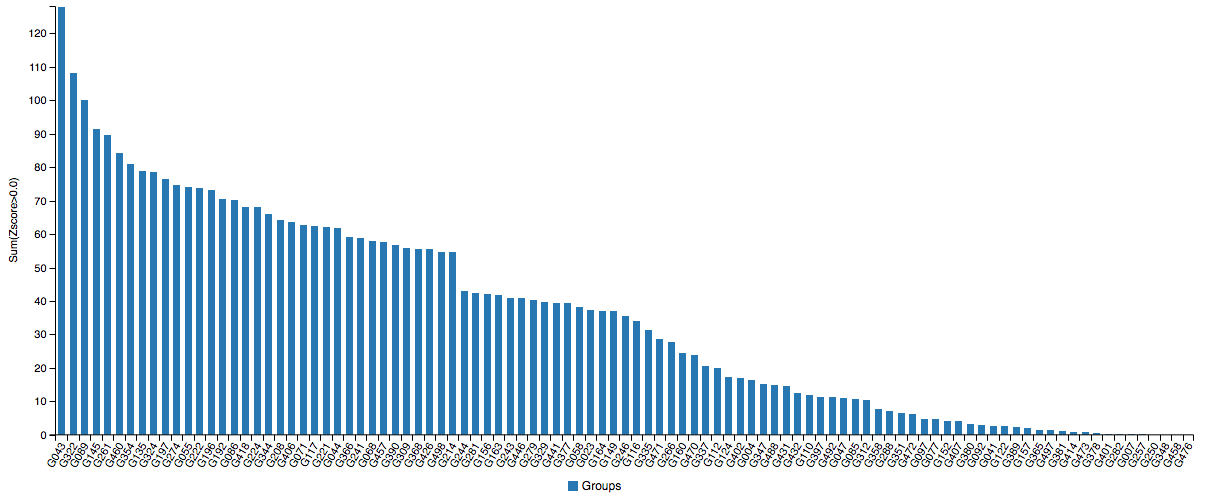 There is only room for one at the top (left-most).
There is only room for one at the top (left-most).
AlphaFold 2: Electric Boogaloo
6.1 Results
Just when you thought AlphaFold was impressive, its sequel blew away everyone else with a 244 sum Z-score. Note that these are single proteins, not categories like multimeric complexes where DeepMind did not participate.
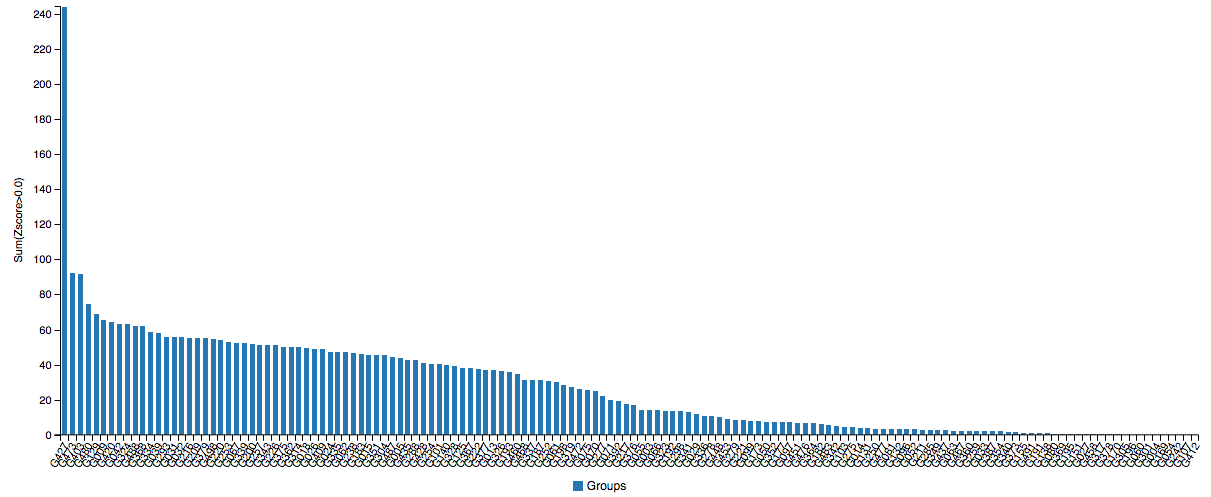 I can’t even see any of you from up here.
I can’t even see any of you from up here.
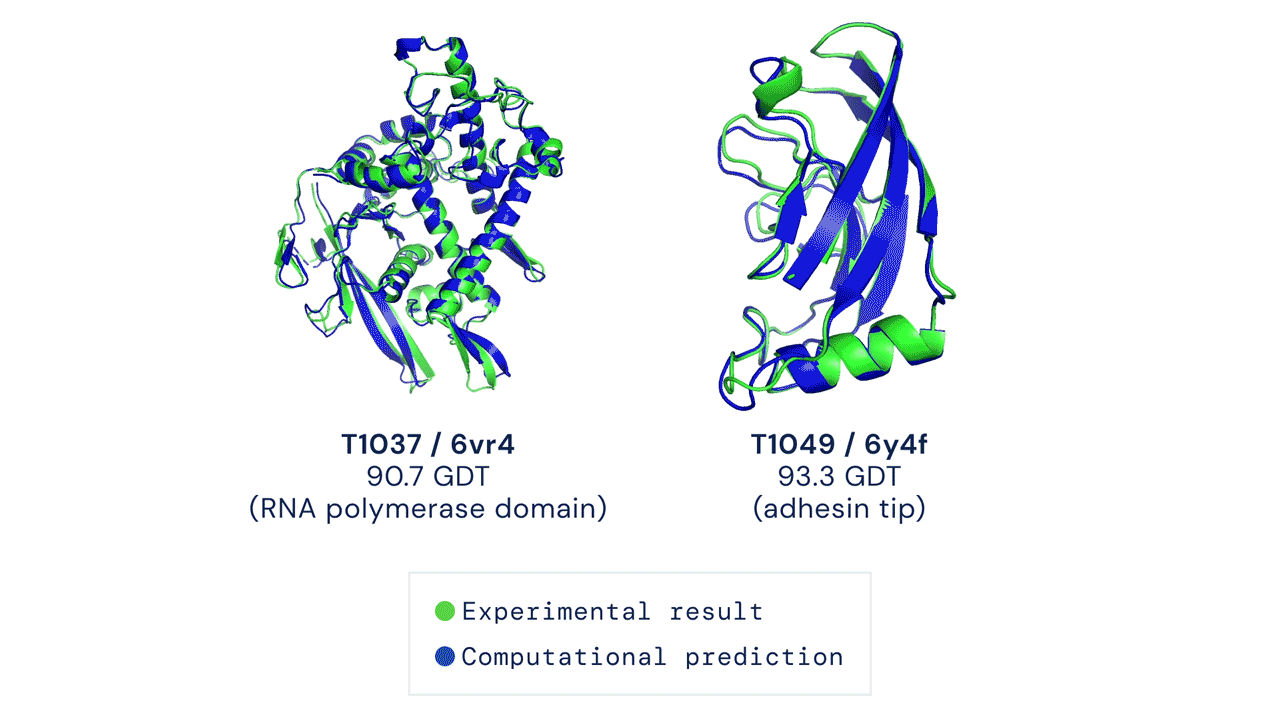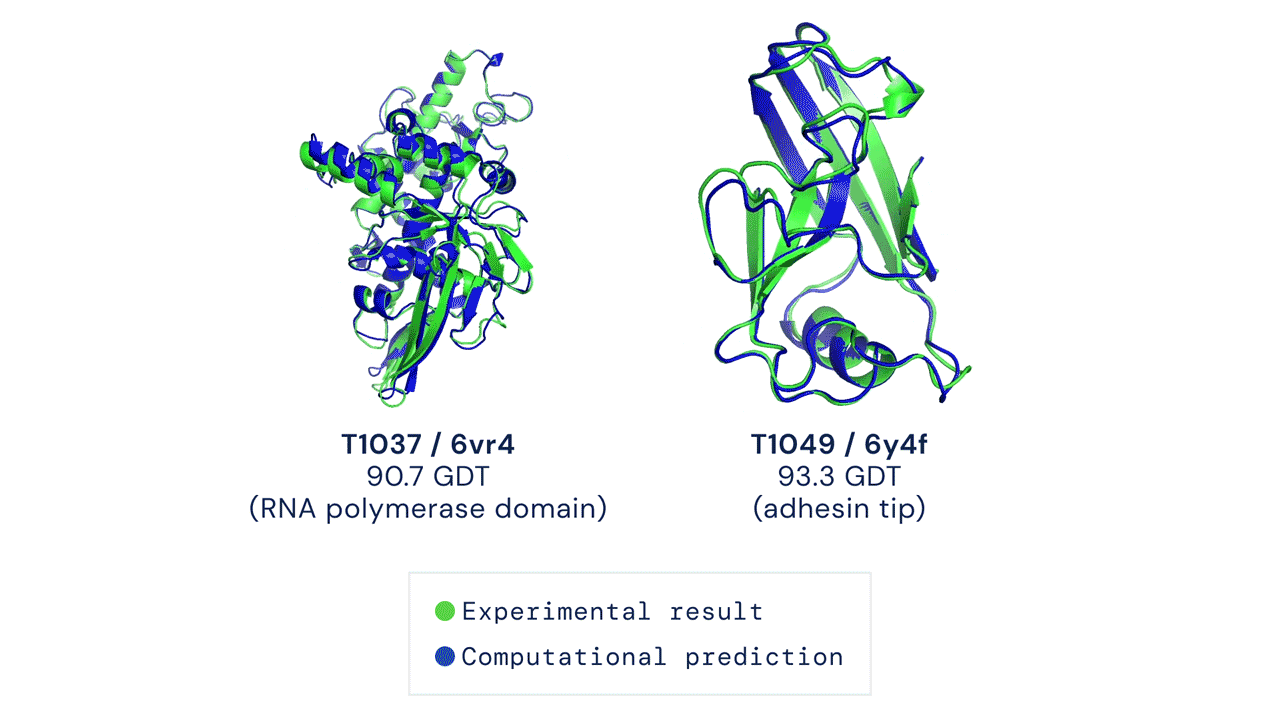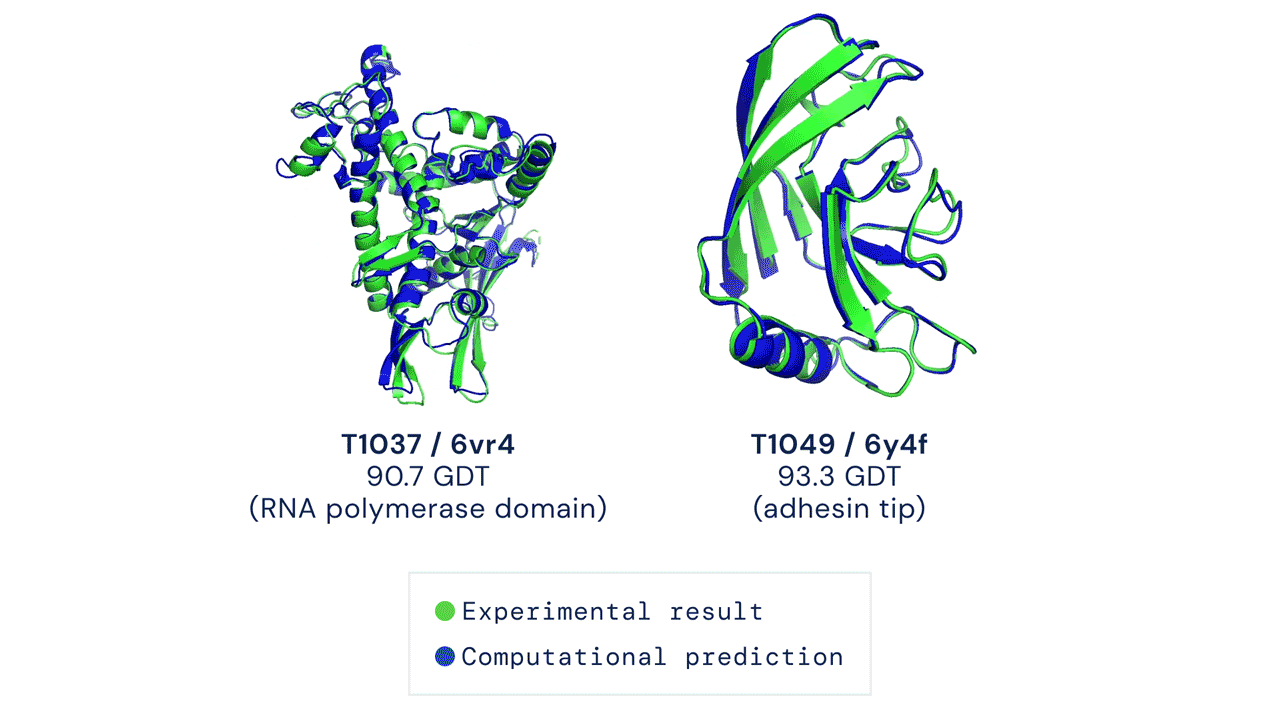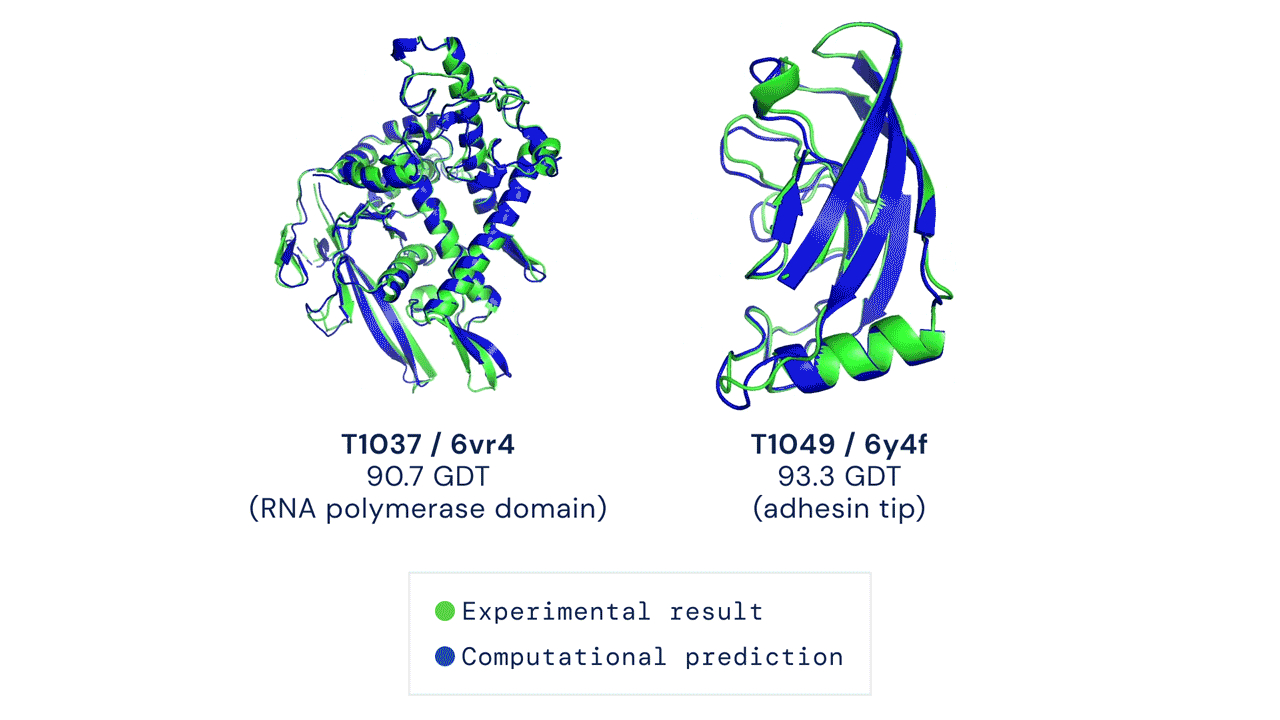
If you think the relative performance is astounding, the improvement in actual performance across the board is nothing short of staggering. AF2 achieved a median score of 92.4 GDT overall across all targets, and a median score of 87.0 GDT in FM. As seen in the graph below, AF2 is able to predict structures with GDT_TS of almost 90 when the next best method only achieves a GDT_TS of 20 (nonsense), and even GDT_TS of above 90 and 95 in some cases when the next best method only achieve a (good) score of about 80.
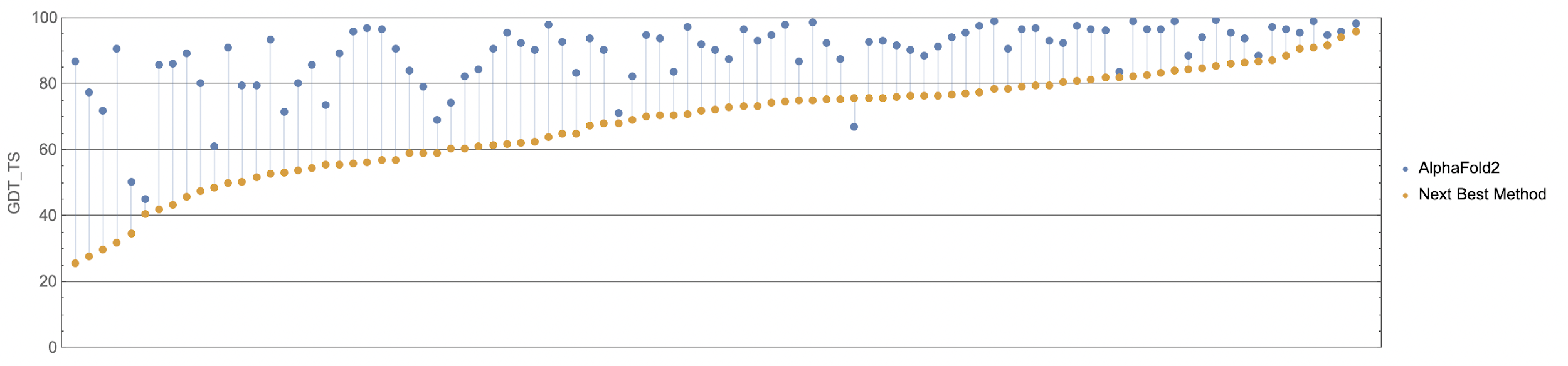 The delta between AF2 and the next best method this year.
The delta between AF2 and the next best method this year.
You can’t deny the magnitude of the breakthrough:
AF2 achieved a mean RMSD of about 1.6Å, and <5Å 92.5% of the time over all side chain atoms. it achieves RMS_CA of <1Å 25% of the time, <1.6Å 50% of the time, and <2.5Å 75% of the time. In terms of RMS_All, it achieves <1.5Å 25% of the time, <2.1Å 50% of the time, and <3Å 75% of the time.
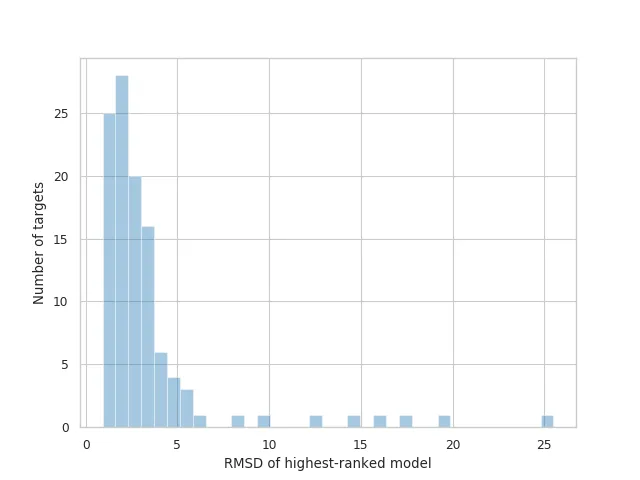
Distribution of RMSDs for the highest-ranked models submitted by AlphaFold 2.
Some results from AF2 are so good that they defied the experimentally determined models. For example, Osnat Herzberg’s group, who were studying a phage tail protein, noticed that they had a different assignment for a cis-proline compared to the model from AF2. Upon reviewing the analysis, they realised they had made a mistake in the interpretation and corrected it. Another example is Henning Tidow’s group, who were trying but struggling to perform X-ray crystallography on an integral membrane protein, reductase FoxB (related to iron uptake in Gram-negative bacteria) for about two years. When they saw the models from AF2, they managed to solve the problem by molecular replacement in a matter of hours.
6.2 A closer look thanks to Carlos Outeiral Rubiera
DeepMind highlighted the ORF8 protein (labelled as T1064 in CASP14; 7JTL in PBD), a viral protein involved with the interaction between SARS-CoV-2 and the immune response, as one of the targets “where they didn’t do that well”.
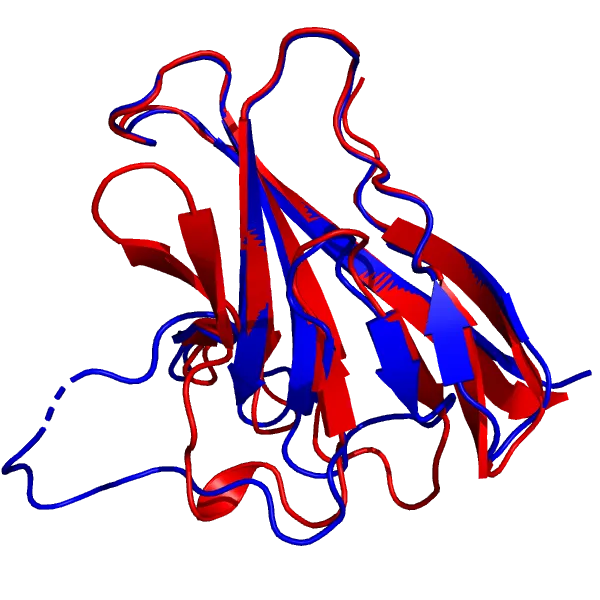
AF2 model for the T1064 target (red), superimposed onto the 7JTL_A structure (blue).
AF2’s model of the core of the protein is in excellent agreement with the experimental structure, down to the antiparallel β-sheets, and more impressively, the loops (top and right) that connect them. As for the large loop region (bottom left) that looks very different from the experimental structure, it does bear some resemblance to the actual loop, and its performance is still better than most common methods. Also, loop regions are commonly very flexible, so it can just mean that that region is mobile. Apparently, a new experimental structure of the ORF8 protein actually displays a much similar structure to the prediction. Loops are generally considered hard to predict, and compared to usual methods, AlphaFold 2’s performance is quite impressive.
How did the Zhang and Baker groups do?
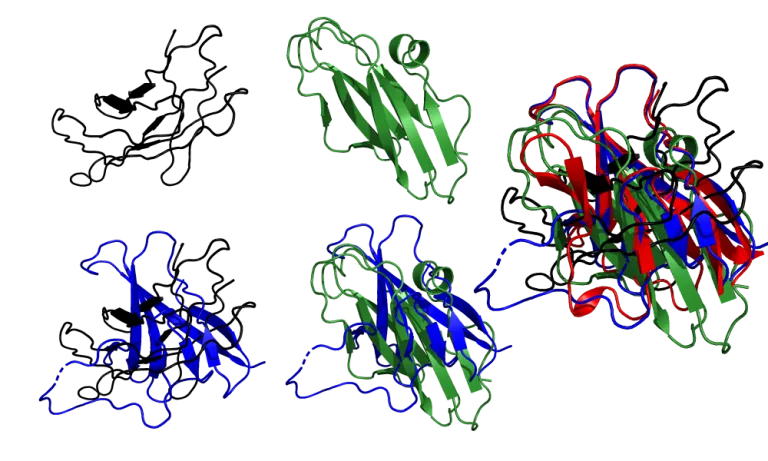
It’s pretty clear that AF2 is in a class of its own. Zhang’s group barely captures the structure of the core, while Baker’s group shows more β-sheets than the crystal structure and the topology falsely combines parallel and antiparallel sheets. In both cases, the loops connecting the β-sheets are all around the place, and the large 30-residue loop region that AF2 didn’t model correctly is modelled even worse by these two submissions.
What about models where DeepMind acknowledges that it did really well? Let’s look at the target T1046s1 (6x6o chain A, in PDB).
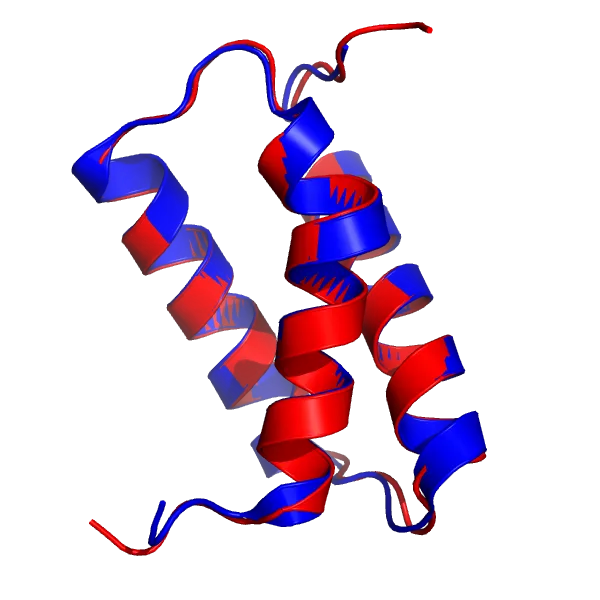
AF2 model for the T1046s1 target (red), superimposed onto the 6X6O_A structure (blue).
The AF2 model, especially the α-helices and the loops, is virtually indistinguishable from the crystal structure with a total RMSD of 0.48Å.
In fact, the agreement is so good that it extends to side chains:
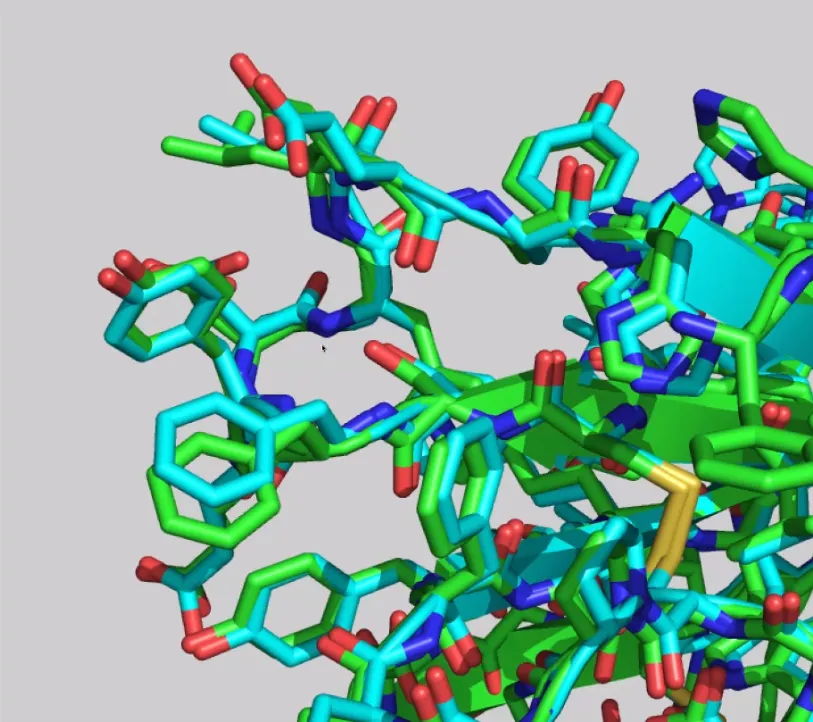
How did the Zhang and Baker groups do?
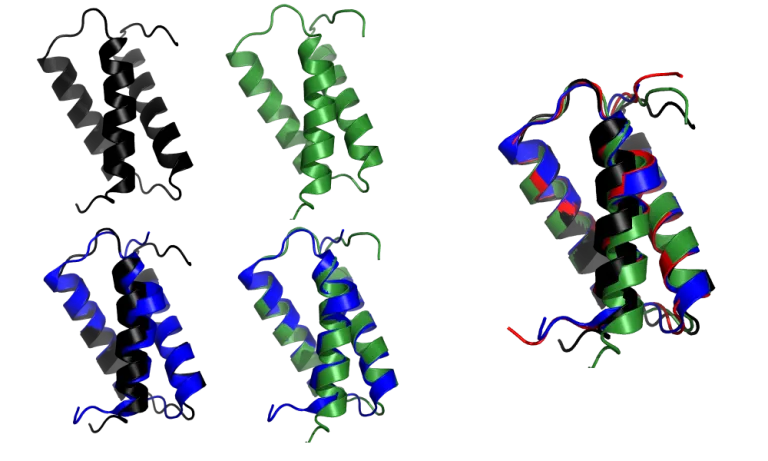
While the Zhang and Baker models are very good, close examination reveals some discrepancies. The kink in the first α-helix is not reproduced accurately: Zhang’s group models it as an essentially straight helix, while Baker groups shows a smaller kink. In comparison, AF2 modelled the kink to perfect accuracy. Also, the magnitude of the deviations is a lot larger than that in the AF2 model.
How Did DeepMind Do It?
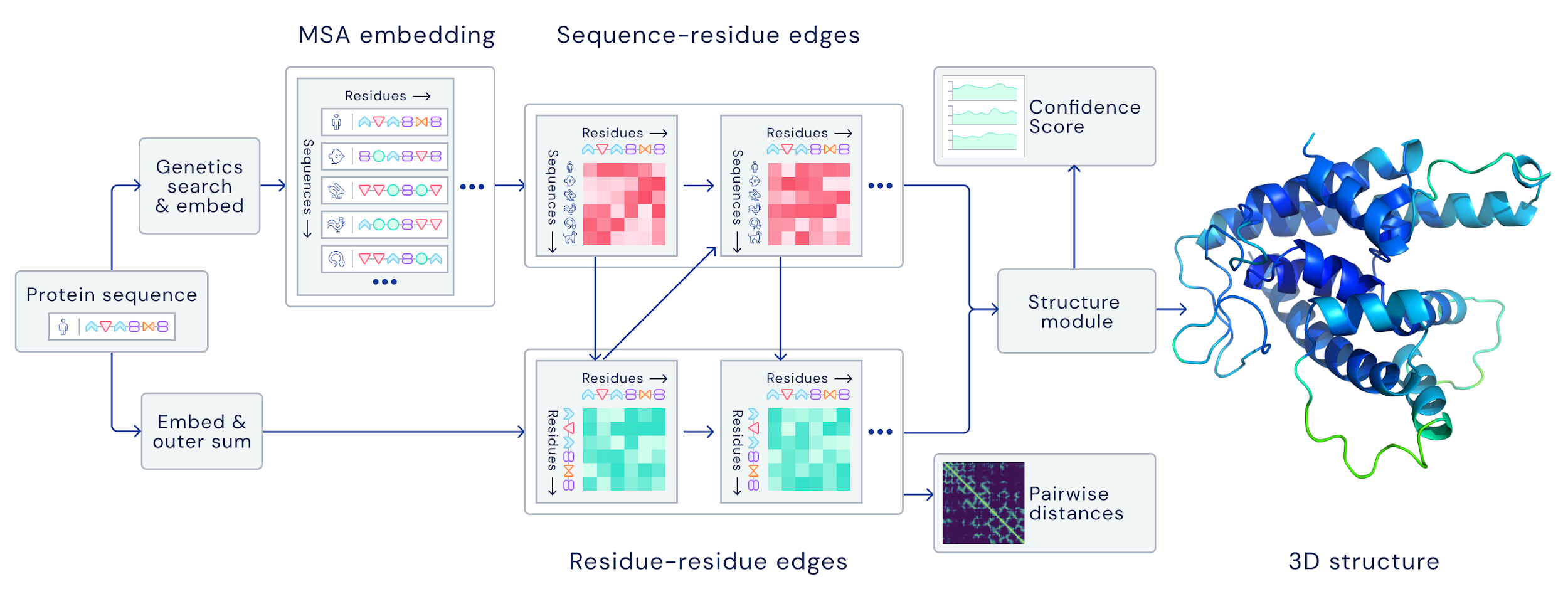
Unfortunately, DeepMind hasn’t published a paper on AF2 yet, so we can only guess.
The current standard (also used by AlphaFold the first) is to extract summary coevolutionary statistics from a multiple sequence alignment (MSA) of homologous protein sequences, then fed into a neural network to predict a “distogram”: a matrix of the probabilities of pairwise distances between all Cβ atoms. This distogram acts as a constraint to narrow the configuration space significantly. MSAs can often be noisy, containing sequences that are not evolutionary related.
DeepMind reformulates the entire pipeline to be end-to-end differentiable; instead of using the MSA to predict distogram constraints, their architecture takes the raw MSA as input and outputs a full structure at the end:
-
Attention model: AF2 decides which raw sequences to look at and which noisy ones to ignore, and from that predicts a distogram, upon which it decides which sequences to attend to next, and so on for a few hundred iterations
-
Leverages deeper (as in having more sequences) MSAs for individual protein domains/intra-domain details, and shallower ones for whole proteins/inter-domain details
-
It doesn’t even explicitly use co-evolution!
-
-
After some number of iterations, AF2 generates a 3D atom cloud from the distograms that is then fed into a transformer for 10 iterations
-
Captures higher-order coordinations between more than 2 atom (information not captured by distograms)
-
It doesn’t use any physics simulation!
-
-
In a few hundred iterations, AF2 begins building the local structure within individual protein domains before branching out to more global features, and outputs a full structure at the end.
- It ensures self-consistency at every step of the pipeline, as traditionally distances between all atoms are first predicted simultaneously (can be nonsense if not embedded in 3D space) then optimised (sometimes physics-based)
-
In TBM, AF2 takes homologues directly as inputs along with the MSA
- But AF2 was also able to perform well in FM
Why DeepMind?
Of course we know that AF2 couldn’t have been invented in a vacuum, but it is especially true as AF2’s attention model uses existing sequences most thoroughly. AF2 is built on all the previous work done by academics, including the whole body of research in peer-reviewed articles, the software tools developed by academic prediction groups, and data painstakingly collected by structural biology groups for decades. Many of the ideas that AF2 incorporates, such as using MSA and templates, all come from the prior research conducted, written up, and reviewed by academics, funded by public money. Also, AF2 is trained using the ~170,000 structures from the PDB and data from the UniRef, as well as data from the BFD and MGnify databases of metagenomics sequences. Moreover, AF2 uses software tools like HHblits, JackHMMER and OpenMM that were developed by academics. DeepMind managed to see further because they stood on the shoulders of giants.
Other than prior work by academics, DeepMind’s success has come as a surprise as no one expected it to come so soon. The novel approach in AF2 and the brilliant engineering can be attributed to 2 reasons why it wasn’t an academic group that solved protein structure prediction.
First, DeepMind has unlimited computing power thanks to unlimited money. Tensor Processing Units (TPUs) are a proprietary Application-Specific Integrated Circuit (ASIC) developed by Google from the ground up for deep learning, unlike GPUs that were originally designed to process graphics. For example, an 8-core TPU v3 chip has 128 GB of vRAM (necessary for high memory-cost attention models like AF2), while an NVIDIA A100 (the GPU with the largest RAM as of writing) “only” has 40 GB. Deepmind says that it uses approximately 16 TPUv3s (which is 128 TPUv3 cores or roughly equivalent to ~100-200 GPUs) run over a few weeks, and renting 128 TPUv2 cores has an annual cost of half a million dollars as per Google Cloud’s pricing page, so AF2’s total computational cost is likely to be in the region of several million dollars. Meanwhile, the Baker and Zhang groups said they used around 4 GPUs for a couple of weeks. This means the AF2 team had roughly two orders of magnitude more computational resources than even the best funded academic research groups.
Interestingly, although DeepMind says that AF2 uses a “relatively modest” amount of compute in the context of most large state-of-the-art machine-learning models, it is in fact an insane amount given that they are not doing any MD.
AlQuraishi says,
“Nothing in their architecture as I understand it could warrant this much compute, unless they’re initializing from multiple random seeds for each prediction. The most computationally intensive part is likely the iterative MSA/distogram attention ping-pong, but even if that is run for hundreds or thousands of iterations, the inference compute seems too much. MSAs can be very large, that is true, but I doubt that they’re using them in their entirety as that seems overkill.”
Vast computational resources doesn’t only allow DeepMind to use mysterious, large models, it also allows a much higher throughput than any academic group. What the Baker group needed a month to test in their 4 Titan GPUs might only take a few hours for DeepMind, allowing for rapid prototyping and testing of ideas.
Second, organisational structure is another key factor beyond unsubstitutable individual contributors. At DeepMind, professionals in the same job with deep expertise spend most of their time doing research, all coordinated in the single direction of building AF2 (which has 18 co-first authors). In academic labs, there are a lot of administrative tasks that take up a lot of time that could’ve been spent doing research, and the constant turnover of students and postdocs means shallower expertise with limited collaboration because at the end of the day it’s about individual effort and building a personal brand. It’s all about the incentives!
However, as AlQuraishi cautions, it would be short-sighted to turn the entire research enterprise into many mini DeepMinds. Unlike protein structure prediction (which literally has a leaderboard every 2 years), most of biology doesn’t have well-defined questions, and the academic model is much better at asking questions, while the DeepMind is only better at answering them. More subtly, the fast and focus model means that there is ironically less time for idea exploration and for new ideas to gestate into the literature to inform questions even beyond protein structure prediction. Once a solution is solved in any way, it becomes hard to justify solving it another way, especially from a publication standpoint. At least for now, we can kind of have our cake and eat it too when fast and focused efforts co-exist with the slow and steady progress of conventional research.
Wait, So Is The Protein Folding Problem Solved?
Again, the protein-folding problem came to be three main questions:
-
The physical folding code: How is the 3D native (properly-folded/functional) structure of a protein determined by the physicochemical properties that are encoded in its 1D amino-acid sequence?
-
The folding mechanism: A polypeptide chain has an almost unfathomable number of possible conformations. How can proteins fold so fast?
-
Predicting protein structures using computers: Can we devise a computer algorithm to predict a protein’s native structure from its amino acid sequence?
Obviously, AF2 has almost no (direct) bearing on the first 2 questions regarding the actual dynamic folding pathways. So I don’t think it’s controversial to say that DeepMind has not solved the protein folding problem per se, but has it solved protein structure prediction? It is itself an umbrella term for distinct problems of predicting:
-
the structure of a single protein domain from sequence,
-
the structure of a single protein, possibly comprised of multiple domains, from sequence,
-
the structure of a multimeric complex,
-
the major conformations of a protein.
For each of the above, there are additional questions:
-
“Purity” of solution: was the prediction made from just a single sequence, or was it made using additional information like homologous protein sequences, homologous structures, and even other forms of non-sequence-based experimental data?
-
Scope of solution: Does a solution need to address all of the corner cases and elaborations e.g. proteins containing metals and other co-factors, unnatural amino acids, entirely de novo proteins?
-
Accuracy: how good is good enough?
Well, AF2 reliably (>90% of the time) predict to reasonable accuracy (<3-4Å) the lowest energy structure of vanilla (no co-factors, no obligate oligomerisation) single protein chains using a list of homologous protein sequences from the PDB (well-known to be biased towards proteins that are easily crystallised).
The list of caveats is a long one, but whether DeepMind solved protein structure prediction can be a bit of a motte (a general solution) and bailey (a universal solution). I think it’s useful to phrase the question as whether we can devise a computer algorithm to predict a protein’s native structure from its amino acid sequence. Since we did not know the answer before AF2 came out, and now we do know that a solution is possible, AF2 counts as a solution, which seems like the consensus among experts like AlQuraishi anyway. The core scientific question of prediction in silico has been answered, so the remaining problems are now engineering rather than scientific (not that engineering problems are any easier or less important).
What Are The Implications?
Mostly a rehash of AlQuraishi’s speculations, do read his essay for more.
Protein structure prediction
AlQuraishi again,
”The core field has been blown to pieces; there’s just no sugar-coating it. I can say this because it’s (one of) my own field(s). There are some intellectually interesting exercises left, for example predicting structure from a single sequence without structural templates or evolutionary information, and there are important engineering problems including addressing all the corner cases that AF2 still can’t. These are important and scientifically worthwhile but will be of limited interest beyond the core community of structure predictors.“
Experimental structure biology
As mentioned above, in the most immediate term, AF2 has already helped X-ray crystallography. However, beyond the short-term (>3-5 years) AF2 will begin to undercut demand for X-ray crystallography as there are a myriad of applications in biology that will benefit from having structures at 3Å or 4Å accuracy that is not as high as X-ray crystallography.
An important caveat is that AF2 is trained on crystallography structures from the PDB, but because cell cytoplasm is not crystal, most crystallized protein structures are probably not particularly good representatives of physiologic state! In the future when other experimental techniques and physics-based computational methods are systematically integrated, we may get predicted structures that would be more informative than crystal structures in determining a protein’s biological function.
What about single particle CryoEM? It has a better short to medium outlook as it is increasingly focused on quaternary complexes and molecular machines, where AF2 can help CryoEM with resolving individual monomers. Still, DeepMind has made it clear that complexes are their next big target, so we will have to see what happens.
The one area that will remain safe and wholly complementary to AF2 is in situ structural biology, because AF2 cannot determine the cellular context of proteins. If anything, AF2 may accelerate the breakneck pace of progress in CryoET and usher in the era of structural cell biology faster than even its proponents are expecting.
Will It Revolutionise Drug Discovery?
No. At least not in the near term.
AF2’s predictions are still not accurate enough to deliver reliable insights into protein chemistry or drug design, which will require accuracy of around 0.3 Å. AF2’s best prediction has an RMSD for all atoms of 0.9 Å. The average RMSD of AF2’s 111 predictions is 1.6 Å (about the size of a bond-length), which is very impressive but still not sub-angstrom.
That said, AF2 might be able to help in determining structures of protein targets that can be modulated for therapeutic purposes designing protein-based (e.g. antibodies and peptides), where ultra-high accuracy is less needed. The challenge is that AF2 is trained to predict apo (unbound) protein structures while most medicinal chemistry applications require complexes of the protein bound to a small molecule.
But in any case, protein structure determination simply isn’t a rate-limiting step in drug discovery in general, not even if X-ray crystallography were to become fast and routine. If you have a solid primary assay against a target you really believe in, and an animal model with real translatability, you don’t need a defined target to get a drug to market. What you absolutely need are safety and efficacy data from successful human clinical trials which require the FDA to allow your IND application. The best way to get an IND approved is to show activity in a relevant animal model and clean toxicology studies in at least two species, and you can do those without knowing the protein target at all, let alone its structure.
The biggest challenge of drug discovery is choosing the wrong target, because our understanding of biology just isn’t there yet, and we can only speculate about how much impact protein structure determination will have on that. So many compounds fail in human trials because turns out our hypothesis about the underlying pathophysiology was just wrong7.
One class of disease where protein structure determination might help us understand better is proteopathy that is caused by protein misfolding.8 Take Alzheimer’s disease for instance, while its exact cause remains unknown, it is associated with toxic aggregations of the misfolded amyloid beta (Aβ) peptide that grows into significantly larger senile plaques. As these aggregates are heterogeneous, experimental methods such as X-ray crystallography and nuclear magnetic resonance (NMR) have had difficulty determining their structures. It doesn’t help that atomic simulations of Aβ aggregation are very computationally demanding due to their size and complexity.
Toxic aggregations of the misfolded Aβ peptide in Alzheimer’s disease.
Another example is prion1, an exception to Anfinsen’s dogma as it is a stable conformation that differs from the native folding state. The misfolding of normal prion protein (PrPc) into prototypical prion disease–scrapie (PrPSc) causes a host of proteopathies known as transmissible spongiform encephalopathies (TSEs) such as Bovine spongiform encephalopathy (Mad Cow Disease), Creutzfeldt-Jakob disease and fatal insomnia. TSEs are especially scary because the ‘seeding’ of the infectious PrPSc, arising spontaneously, hereditary or via contamination, can cause a chain reaction of transforming normal PrPc into fibrils aggregates or amyloid like plaques consisting of PrPSc. The molecular structure of PrPSc has not been fully characterised due to its aggregated nature. Neither is known much about the mechanism of the protein misfolding nor its kinetics.
AF2 may give us some insights into proteopathies if we can analyse how the neural network infers the folded structure, but that can be even harder than protein folding (neural nets are black boxes), and even if we manage to it might also be useless simply because the network’s inference is just not very representative of the dynamic folding process.
Better models (animal and otherwise) of such diseases and conditions would be helpful too, but that also depends on how much we know about underlying pathophysiology. For example, the underlying difficulty of countless compounds for Alzheimer’s that work in animal models but fail in human trials is that humans are the only animal that actually gets Alzheimer’s. It’s a chicken-and-egg question; you would need to know a lot more about the disease before you can create a good model.
In the long run, the true power of AF2 may come in providing a more robust platform for discovery drugs based on their polypharmacology i.e. to modulate multiple protein targets intentionally, which may be able to modulate entire signaling pathways instead of acting on one protein at a time. There’s a lot more work to be done, but in the meantime, we can appreciate that AF2 is one of the most significant scientific breakthroughs in our lifetime.
Endnotes
-
One of Eleizer Yudkowsky’s scenarios of AI takeover:
-
Crack the protein folding problem to the extent of being able to generate DNA strings whose folded peptide sequences fill specific functional roles in a complex chemical interaction.
-
Email sets of DNA strings to one or more online laboratories that offer DNA synthesis, peptide sequencing, and FedEx delivery.
-
Find at least one human connected to the Internet who can be paid, blackmailed, or fooled by the right background story, into receiving FedExed vials and mixing them in a specified environment.
-
The synthesized proteins form a very primitive “wet” nanosystem, which, ribosome-like, is capable of accepting external instructions; (…)
-
Use the extremely primitive nanosystem to build more sophisticated systems, which construct still more sophisticated systems, bootstrapping to molecular nanotechnology—or beyond.
-
-
Proteins are chains of amino acids joined together by peptide bonds. The many 3D conformations of this chain are possible due to the rotation of the chain about each alpha carbon (Cα) atom, the backbone carbon before the carbonyl carbon atom in the protein molecule. So what forces drive the rotation of the amino acid chains? Some are:
-
Hydrogen bonds: α-helix and β-sheet were predicted by Linus Pauling from hydrogen-bond models
-
van der Waals interactions: duh
-
Backbone angle preferences: polymers have preferred angles of neighbouring backbone bond orientations
-
Electrostatic interactions: some amino acids attract or repel because of negative and positive charges
-
Hydrophobic interactions: proteins ball up with the hydrophobic amino acids in the core and the polar amino acids on the surface
-
Chain entropy: opposing the folding process is a large loss in chain entropy as the protein collapses into its compact native state from its many open denatured configurations
Through statistical mechanical modelling in the 1980s, minimising the number of hydrophobic side-chains exposed to water, or the hydrophobic effect, has been shown to be the predominant driving force behind the folding process. An ordering of water molecules around a hydrophobic region (“water cage”) contributes a negative change in entropy. The hydrophobic collapse (inward folding of the hydrophobic groups) introduces entropy back to the system via the breaking of the “water cages” which frees the ordered water molecules. The multitude of hydrophobic groups interacting within the core of the folded protein significantly contributes to protein stability after folding due to the vastly accumulated van der Waals forces.
Currently, we don’t fully understand the energetic processes and tradeoffs in folding any given protein, but this field of energetics in protein folding is advancing. At the end of the day, we still haven’t found sequence patterns, say of hydrophobic, polar, charged, and aromatic amino acids, that would predict protein structures and stabilities. So the first question of the protein folding problem remains unsolved. ↩
-
-
There have been many attempts to get faster at doing these kinds of calculations. IBM’s (retired?) Blue Gene supercomputers boasted of operating speeds in the petaFLOPS range with lower power consumption, but I couldn’t find any protein prediction paper from them at all, and it seems that they have ended development in 2015. ↩
-
Anfinsen’s dogma (a.k.a. the thermodynamic hypothesis) postulates that the native structure of a protein is its most thermodynamically stable structure; it depends only on the amino acid sequence and on the conditions of solution, and not on the kinetic folding route.
Keeping in mind that systems tend to move towards its global free energy (energy available in a system to do useful work) minimum, there are three conditions:
-
Uniqueness: the free energy minimum must be unchallenged; the sequence does not have any other configuration with a comparable free energy
-
Stability: small changes in the surrounding environment cannot give rise to changes in the minimum configuration; the free energy surface around the native state must be rather steep and high, in order to provide stability
-
Kinetical accessibility: the path in the free energy surface from the unfolded to the folded state must be reasonably smooth; the folding of the chain must not involve highly complex changes in the shape (e.g. knots)
Put simply, to determine a protein’s native structure, all the information you need is in its amino-acid sequence2,3. But at the end of the day, we still haven’t found sequence patterns, say of hydrophobic, polar, charged, and aromatic amino acids, that would predict protein structures and stabilities. So the first question of the protein folding problem remains unsolved.
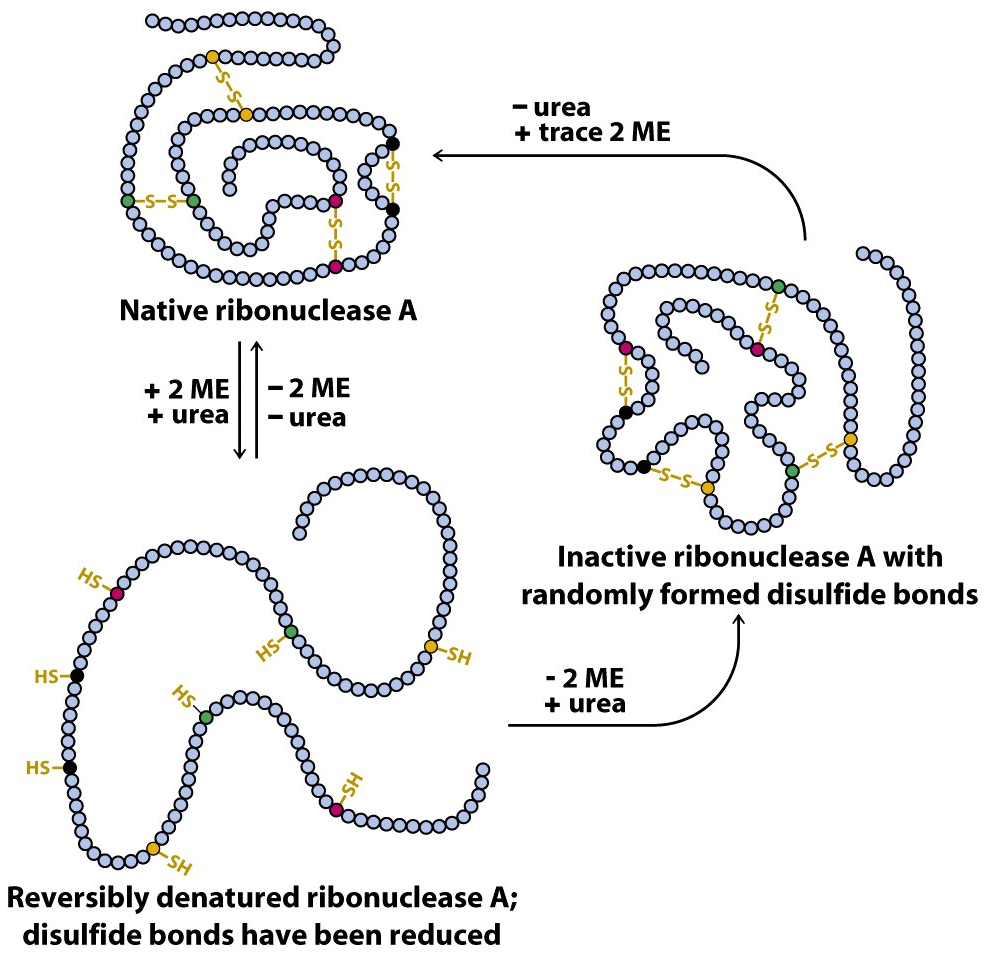
Anfinsen demonstrated it in his now-famous experiments from 1960 to 1962 by using a protein called ribonuclease A (RNase A), which contains four disulfide bridges that are vital to protein structure and function. First, he unfolded RNase A with urea (which denatures or causes the loss of native structure of protein) and βME (a reducing agent that breaks disulfide bridges). When Anfinsen removed urea first and βME second, the protein refolded and regained 100% activity. However, when he removed βME first and urea second, the protein only regained 1% of its original activity. This was because removing βME when the enzyme is still unfolded causes cysteine (an amino acid) residues to randomly form disulfide bonds with each other.
There are some caveats. Some proteins need the assistance of another protein called a chaperone protein which seems to work primarily by preventing aggregation of several protein molecules prior to the final folded state of the protein. It has been suggested that this disproves Anfinsen’s dogma, but many chaperones do not appear to affect the final state of the protein. ↩
-
-
See my post about Folding@home as a scientific lemon project running on high resource costs and why charity is still not about helping. ↩
-
Another thing that will speed up drug discovery is a better warning system for toxicity in human trials as well. Many promising drugs have dropped out of the clinic due to unexpected tox effects, for sure – some of these turn out to be mechanism-related and some of them are just compound-related (where the compound does something else that you don’t want), but there are many instances where we can’t even make that distinction yet. Animal models for toxicity are extremely valuable, but they don’t get you all the way. You are still taking a risk every time a new compound or new mechanism goes into human trials, and it would be very useful if we could lower that risk a bit. The general solution would be some sort of system that exactly mimics human biology but doesn’t consist of a bunch of human swallowing pills. This is a difficult goal to realize. ↩
-
In 2011, the European Medicines Agency approved the use of Tafamidis a.k.a. Vyndaqel/Vyndamax (a kinetic stabilizer of tetrameric transthyretin) to delay peripheral nerve impairment in adults with transthyretin amyloid diseases. In 2019, the FDA approved Vyndaqel and Vyndamax for the treatment of transthyretin mediated cardiomyopathy. This suggests that the process of amyloid fibril formation (and not the fibrils themselves) causes the degeneration of post-mitotic tissue in human amyloid diseases. ↩